|
Patrick J. Desrochers UCA webpage |
Research Interests · promoting
undergraduate research · scorpionates ligands anchored to polymer supports · phosphine-nickel-cysteine complexes · chalcogen selectivity by
nickel · atypical nickel-cysteine centers · reversible alkylation of nickel-cysteine centers
· transition metal
borohydrides · reversible
nickel-ammine binding · electronically
modified Tp* |
Links for current research students responsible
conduct for research (RCR) plan “The Lab” research ethics simulation (Dept. of HHS)
This work has been supported by ·
National
Science Foundation (CHE 0717213) and ·
ACS
Petroleum Research Fund (35602-B3
and 39644-B3) |
|
Routine Paramagnetic 11B NMR from NMR Spectroscopy in the Undergraduate
Curriculum ACS Symposium
Series v. 1128; American Chemical Society: Washington, DC, 2013, Ch7, p
182. DOI:
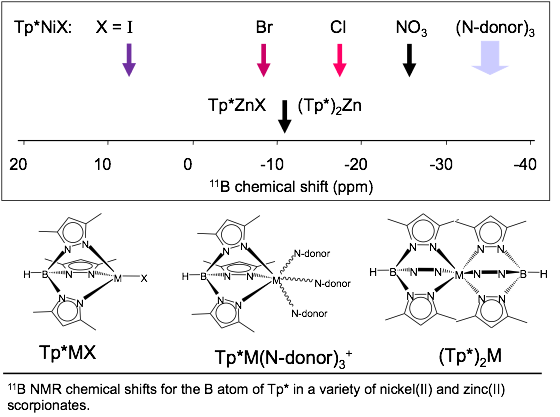
10.1021/bk-2013-1128 The boron heteroatom in paramagnetic Tp*NiX (X = Cl,
Br, I, NO3, BH4, or a second TpR)
provided undergraduate researchers the opportunity to develop a unique 11B
NMR chemical shift range that was very sensitive to both the
identity of X and the metal coordination geometry. The influence of the nickel(II)
paramagnet (S = 1) on the Tp* boron atom is clearly evident. Typical through-space nickel-boron
distances in Tp*NiX are
~3 Å (X = Cl, Br, BH4). Chemical
shifts for four-coordinate Tp*NiX
geometries vary widely with X. Five-coordinate Tp*NiX geometries (or fluxional variations) give chemical
shifts near -25 ppm and six-coordinate octahedral Tp*NiX cases give chemical shifts near -36 ppm over a wide
range of N-donors (including a second Tp*, 3 NCCH3,
or 3 NH3). In contrast the
boron chemical shift in the closed-shell zinc(II)
cases, Tp*ZnX, is
insensitive to X, such that the boron chemical shifts of Tp*ZnX (X= Cl and I) and Zn(Tp*)2 only differ by 0.5 ppm. The broad utility of scorpionates in
paramagnetic transition metal complexes motivates development of meaningful
B-11 chemical shift libraries of these complexes. The nickel(II)
examples demonstrate the usefulness of this method for rapidly identifying
metal-scorpionate coordination geometries in reaction mixtures. The
combination of such trends and the general excellent sensitivity of 11B
has made this tool a quick and routine confirmatory measurement for existing
and newly prepared metal scorpionates in our work. For example, this trend was useful in
identifying in situ formation of
Tp’NiNO3 (11B d ~
25 ppm), a product of this new heteroscorpionate but one that proved
difficult to isolate from reaction mixtures |
|
|
|
|
|
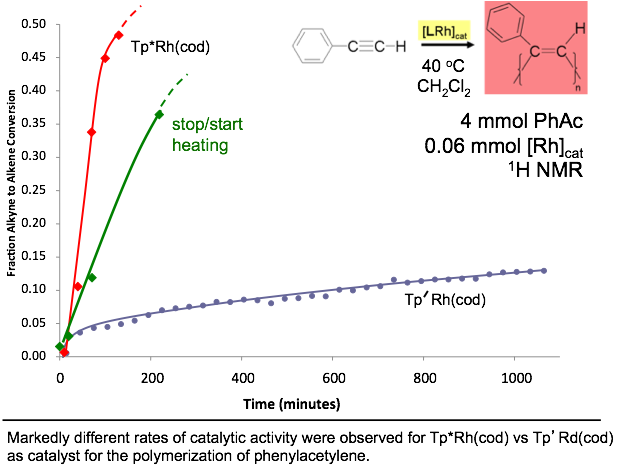
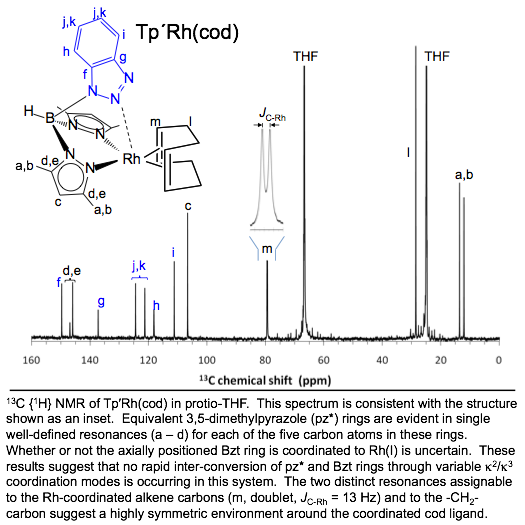
A Rhodium(I)
Heteroscorpionate Catalyst for Phenylacetylene Polymerization Rhodium(I) scorpionates have demonstrated activity in the polymerization of phenylacetylene, a bright orange conjugated polymer with useful electrical and optical properties. Recently we reported the preparation of scorpionates (Tp’) anchored to polystyrene synthesis beads (bead-Tp’ in Inorg. Chem. 2011, p. 1931). These supported chelates offer the versatility of scorpionates to rapid-throughput combinatorial methods. This motivation led to the preparation of Tp’Rh(cod), where Tp’ represents the new tridentate chelate, hydrobis(3,5-dimethylpyrazolyl)(benzotriazolyl)borate. The catalytic activity of Tp’Rh(cod) toward phenylacetylene polymerization was compared to the established analogue Tp*Rh(cod) (Tp* = hydrotris(3,5-dimethylpyrazolyl)-borate). Marked differences in catalytic activity of these two complexes are ascribed to the variable hapticity (k2 vs k3) preferences of the two scorpionates. Completely different rates of activity were also noted when a more electron-rich monomer (p-H3C-PhC≡CH) replaced phenylacetylene. The present results for homogeneous samples of Tp’Rh(cod) will help describe subsequent activity studies in heterogeneous supported systems, bead-Tp’Rh(cod). Here, the brightly colored polymer-product will allow rapid optical screening of favorable supported-catalyst candidates. |
|
|
|
|
|
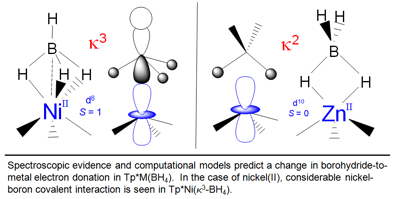
Electronic Structure of Nickel(II)
and Zinc(II) Borohydrides from Spectroscopic Measurements and Computational
Modeling Inorganic Chemistry 2012, 51, 2793-2805. DOI: 10.1021/ic201775c. The previously reported Ni(II)
complex, Tp*Ni(k3-BH4)
(Tp* = hydrotris(3,5-dimethylpyrazolyl)borate
anion), which has an S = 1 spin
ground state, was studied by high-frequency and -field electron paramagnetic
resonance (HFEPR) spectroscopy as a solid powder at low temperature, by UV-Vis-NIR
spectroscopy in the solid state and in solution at room temperature, and by
paramagnetic 11B NMR. HFEPR
provided its spin Hamiltonian parameters: D
= 1.91(1) cm-1, E =
0.285(8) cm-1, g =
[2.170(4), 2.161(3), 2.133(3)]. Similar, but not identical parameters were
obtained for its borodeuteride analog. The
previously unreported complex, Tp*Zn(k2-BH4),
was prepared and IR and NMR spectroscopy allowed its comparison with
analogous closed shell borohydride complexes. Ligand-field theory was used to
model the electronic transitions in the Ni(II)
complex successfully, although it was less successful at reproducing the
zero-field splitting (zfs) parameters. Advanced computational methods, both
density functional theory (DFT) and ab initio wavefunction
based approaches, were applied to these Tp*MBH4
complexes to better understand the interaction between these metals and
borohydride ion. DFT successfully reproduced bonding geometries and
vibrational behavior of the complexes, although it was less successful for
the spin Hamiltonian parameters of the open shell Ni(II)
complex. These were instead best described using ab initio methods. The origin of the zfs in Tp*Ni(k3-BH4)
is described and shows that the relatively small magnitude of D results from several spin-orbit
coupling (SOC) interactions of large magnitude, but with opposite sign.
Spin-spin coupling (SSC) is also shown to be significant, a point that is not
always appreciated in transition metal complexes. Overall, a picture of
bonding and electronic structure in open and closed shell late transition
metal borohydrides is provided, which has
implications for the use of these complexes in catalysis and hydrogen
storage.
|
|
|
|
|
|
Coordination behavior of a new heteroscorpionate toward
second-row transition metals |
Spectral
evidence supports the coordination behavior of Tp′ shown with
molybdenum(0) and rhodium(I) below. Clean room
temperature NMR measurements suggest no rapid interconversion of Bzt and
pyrazole rings is occurring
in these systems. |
|
|
|
|
|
|
|
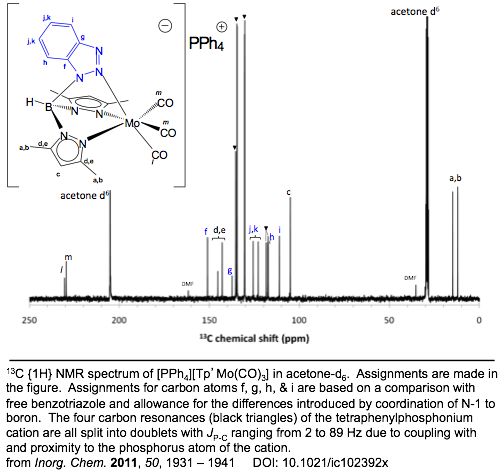
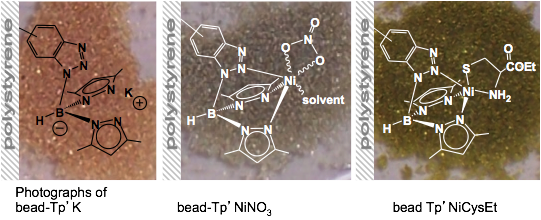
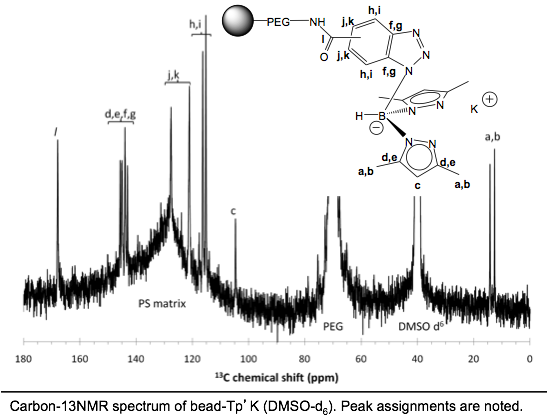
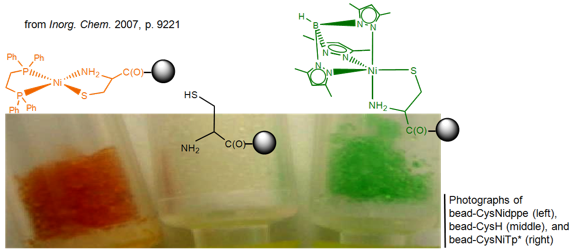
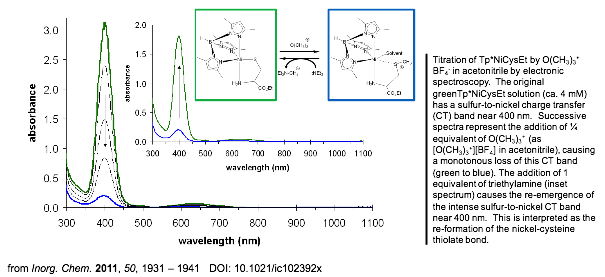
Boron-scorpionates anchored to polymer supports Inorganic Chemistry 2011,
50, 1931 – 1941. DOI: 10.1021/ic102392x The
preparation of a resin-supported boron-scorpionate ligand and its nickel(II) coordination
complexes are reported. The supported ligand is prepared as its potassium
salt, making it a general reagent suitable for chelation of any transition
metal ion. Resin-immobilized benzotriazole (Bead-btz) reacted cleanly with KTp* (Tp* = hydrotris(3,5-dimethylpyrazolyl)borate) by
heterocycle metathesis in warm dimethylformamide (DMF) to
yield bead-Tp’K,
{resinbtz(H)B(pz*)2}K.
Significantly, bead-Tp’K readily bound nickel(II) from
simple salts with minimal leaching of the nickel ion. Bead-Tp’NiNO3
reacts further with cysteine thiolate (ethyl
ester), imparting the deep green color to the beads characteristic of a
TpRNiCysEt coordination sphere. Bead-Tp’NiCysEt
exhibited an oxygen sensitivity similar to Tp*NiCysEt in solution (Inorg. Chem. 1999, p 5690) and
also independently verified for a selenocystamine
analogue, Tp*NiSeCysAm.
Addition of fresh cysteine thiolate ethyl ester to oxidized bead-Tp’NiCysEt
reproduced the original green color. Heterocycle metathesis was also used to
prepare KTp’ as
a white solid. Reaction with nickel(II) gave (Tp’)2Ni,
separable into two different isomers. The air-sensitive molybdenum(0) complex,
[PPh4][Tp’Mo(CO)3],
was also prepared and the Cs
complex symmetry demonstrated by infrared and 13C NMR
spectroscopies. Immobilized TpmMo(CO)3
was prepared from the previously reported resin-supported tris(pyrazolyl)methane.
In contrast to its weak coordination of nickel(II) (Inorg. Chem. 2009, p 3535),
bead-Tpm proved a strong chelate toward this second
row metal. The supported scorpionates described here should find use in
studies of selective metal-protein binding, metalloprotein modeling, and
heterogeneous catalysis, and render such scorpionate applications amenable to
combinatorial methods.
|
|
|
|
|
|
|
|
|
Polymer supported tris(pyrazolyl)methane,
minimum steric constraints Inorganic Chemistry 2009, 48, 3535-3541 DOI: 10.1021/ic8015645 Single-scorpionates of nickel(II),
TpRNiX or TpmRNiX, are kinetic
products whose preparation has generally required considerable steric constraints on the
ligands (i.e., R ) phenyl, tert-butyl, or
isopropyl) to prevent formation of intractable two-ligand products like (TpR)2Ni. It is well established that the facial
tridentate chelates hydrotris(3,5- dimethylpyrazolyl)borate (Tp*-), tris(3,5-dimethylpyrazolyl)methane (Tpm*),
and trispyrazolylmethane (Tpm),
all readily form two-ligand complexes as thermodynamic products. For the first
time we report a route to the single-ligand complex TpmNiX2(OH2)n (X ) Cl and Br).
We also report a novel method for making single-ligand nickel(II) scorpionate complexes using preformed tetrahalonickelate(II)
ion in nitromethane. The complex Tpm*NiCl2(OH2)n
was also prepared here for the first time utilizing an alternative method first
reported by Zargarian and co-workers (Inorg. Chim. Acta 2006, 2592). TpmNiX2(OH2)n are kinetic products, and although
they are stable indefinitely in the solid state, they readily convert to the thermodynamic product
(Tpm)2Ni2+ in solution over
the course of several hours at room temperature and in a matter of minutes at 100 °C. The
new nitromethane/NiX42- method offers an alternative
route to monoscorpionates of first row
transition metals, for which tetrahalometallate
ions are common. HOCH2Tpm (2,2,2-tris(pyrazolyl)ethanol) was covalently
attached to polystyrene synthesis beads and found to bind nickel(II) (from NiX42-) in a
manner similar to Tpm. Solid state electronic
spectra of supported-TpmNiCl2 are comparable to those measured for their homogeneous complexes. Covalently supported
scorpionates are expected to further extend the utility of this rich ligand class in
areas of heterogeneous catalysis and metal-protein interactions. |
|
|
|
|
|
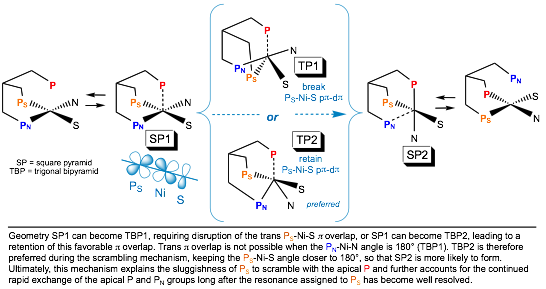
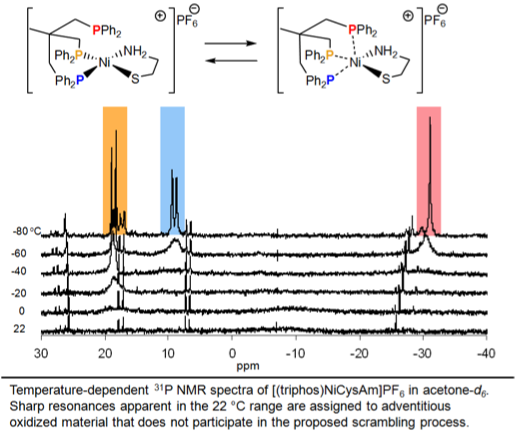
Phosphine-nickel-cysteine
complexes Inorganic Chemistry 2007,
46, 9221 - 9233 DOI: 10.1021/ic701150q The effect of chelating phosphines
was tested on the structure and pH-dependent stability of nickel-cysteine
binding. (1,2-Bis(diphenylphosphino)ethane
(dppe) and 1,1,1-tris[(diphenylphosphino)methyl]ethane
(triphos) were used with three different cysteine derivatives (L-cysteine, Cys; L-cysteine ethyl ester, CysEt; cystamine, CysAm) to prepare complexes of the form (dppe)NiCysRn+ and (triphos)NiCysRn+ (n = 0 for Cys; n =
1 for CysEt
and CysAm). Similar 31P {1H} NMR spectra for all (dppe)NiCysRn+ confirmed
their square-planar P2NiSN coordination spheres. The structure of [(dppe)NiCysAm]PF6 was also confirmed by single-crystal X-ray diffraction methods.
The (triphos)- NiCysAm+ and (triphos)NiCysEt+ complexes
were fluxional at room temperature by 31P NMR. Upon cooling to -80 °C, all gave spectra consistent with a P2NiSN coordination sphere with the third phosphorus uncoordinated. Temperature-dependent 31P NMR
spectra showed that a trans P-Ni-S p interaction
controlled the scrambling of the coordinated triphos. In aqueous media,
(dppe)NiCys was
protonated at pH ~ 4-5, leading to possible formation of a nickel-cysteinethiol and eventual
cysteine loss at pH < 3. The importance of N-terminus cysteine in such complexes was demonstrated by preparing (dppe)NiCys-bead and
trigonal-bipyramidal Tp*NiCys-bead
complexes, where Cys-bead represents cysteine anchored
to polystyrene synthesis beads and Tp*-
) hydrotris(3,5- dimethylpyrazolyl)borate. Importantly, results with these
heterogeneous systems demonstrated the selectivity of these nickel centers for cysteine over methionine and serine and most
specifically for N-terminus cysteine. The role of Ni-S pi bonding in nickel-cysteine geometries will
be discussed, including how these results suggest a mechanism for the movement of electron density from
nickel onto the backbone of coordinated cysteine.
|
|
|
|
|
|
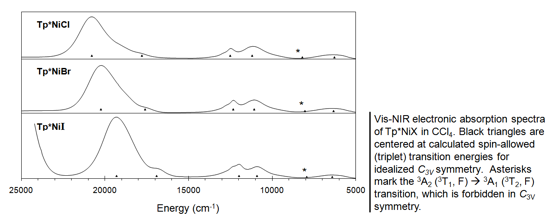
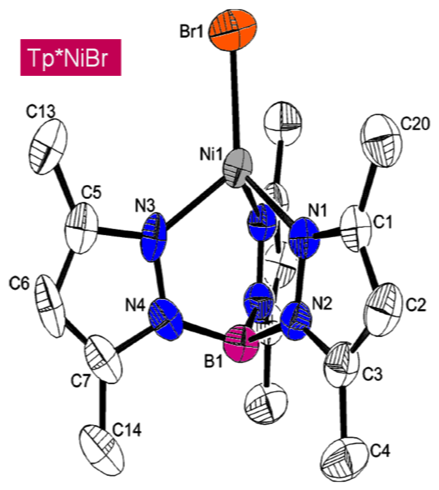
Electronic structure of half-sandwich
nickel(II)-scorpionates Inorganic Chemistry 2006, 45, 8930 - 8941 DOI: 10.1021/ic060843c A series of complexes of formula Tp*NiX, where Tp*- )
hydrotris(3,5-dimethylpyrazole)borate and X = Cl,
Br, I, has been characterized by electronic
absorption spectroscopy in the visible and near-infrared (NIR) region and by high-frequency and -field electron paramagnetic resonance (HFEPR)
spectroscopy. The crystal structure of Tp*NiCl has been previously reported; that for Tp*NiBr is given here: space
group = Pmc21, a =
13.209(2) Å, b = 8.082(2) Å, c )
17.639(4) Å, a
= b =
g = 90°, Z = 4. Tp*NiX
contains a four-coordinate nickel(II) ion (3d8) with approximate C3v point group symmetry about the
metal and a resulting S = 1 high-spin ground state. As a consequence of sizable zero-field splitting
(zfs), Tp*NiX complexes
are “EPR silent” with use of conventional EPR; however, HFEPR allows observation of multiple transitions. Analysis of
the resonance field versus the frequency dependence of these transitions allows extraction of
the full set of spin Hamiltonian parameters. The axial zfs parameter for Tp*NiX displays pronounced halogen contributions down the
series: D = +3.93(2), -11.43(3), -22.81(1) cm-1, for
X = Cl, Br, I, respectively. The magnitude and change in sign of D observed
for Tp*NiX reflects the increasing bromine and iodine spin-orbit contributions
facilitated by strong covalent interactions with nickel(II). These spin Hamiltonian parameters are
combined with estimates of 3d energy levels based on the visible-NIR spectra to yield ligand-field parameters for these complexes following
the angular overlap model (AOM). This description of electronic structure and
bonding in a pseudotetrahedral nickel(II) complex
can enhance the understanding of similar sites in metalloproteins, both
native nickel enzymes and nickel-substituted zinc enzymes.
|
|
|
|
|
|
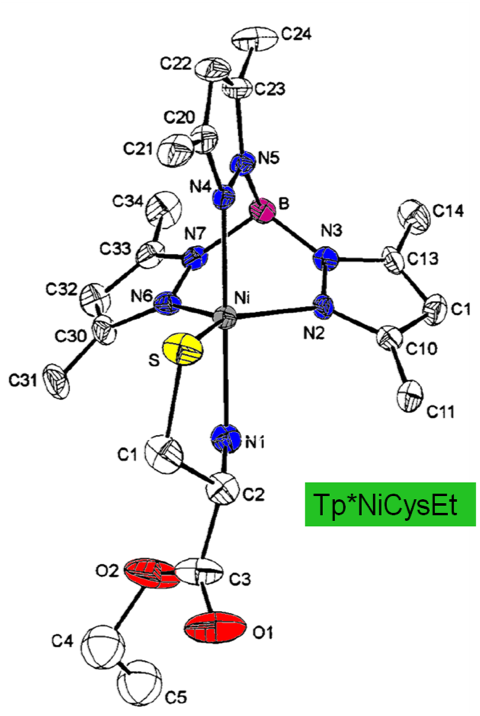
Nickel-cysteine selectivity Inorganic Chemistry 1999, 38, 5690 - 5694 DOI: 10.1021/ic990059a Monomeric five-coordinate nickel-cysteine
complexes were prepared using anionic tris(3,5-disubstituted pyrazolyl)- borates (Tp* - and TpPhMe-) and l-cysteine (ethyl ester and amino acid forms). Tp*NiCysEt crystallizes with a single methanol of solvation in the
monoclinic space group P21: a
= 7.8145(18), b = 24.201(6), c = 7.9925(14) Å; b = 117.991(16)°. [Tp*NiCys-][K+] and TpPhMeNiCysEt show magnetic and
electronic characteristics similar to Tp*NiCysEt, so that the trigonal bipyramidal coordination
geometry confirmed for Tp*NiCysEt
in the solid state likely applies to all three. All three complexes have high spin
magnetic ground states at room temperature (meff )
2.9-3.2 mB, S =1). Their electronic spectra are dominated
by sulfur to nickel charge-transfer bands (388- 430 nm in chloroform) with energies that
correlate to respective thiolate basicities and TpX- donor
strengths. The Tp* derivatives undergo a rapid reaction with molecular oxygen. Stoichiometric,
infrared, and electronic spectroscopy measurements are consistent with formation
of a sulfinate as a result of reaction with dioxygen. Kinetics measurements for the reaction of Tp*NiCysEt and O2 fit the following composite rate law: rate = k1[Tp*NiCysEt] + k2[O2][Tp*NiCysEt] with k1 =
0.013(1) min-1 and k2 =
4.8(1) M-1 min-1 at 22 °C. Increased nucleophilicity of the nickel-sulfur center enhanced by
electron donation from Tp*- (vs TpPhMe-) and encouraged by a trigonal bipyramidal geometry (vs square planar Ni(CysEt)2) is hypothesized as the reason for the susceptibility of Tp*NiCys complexes to oxygen.
|
|
|
|
|
|
|
|
|
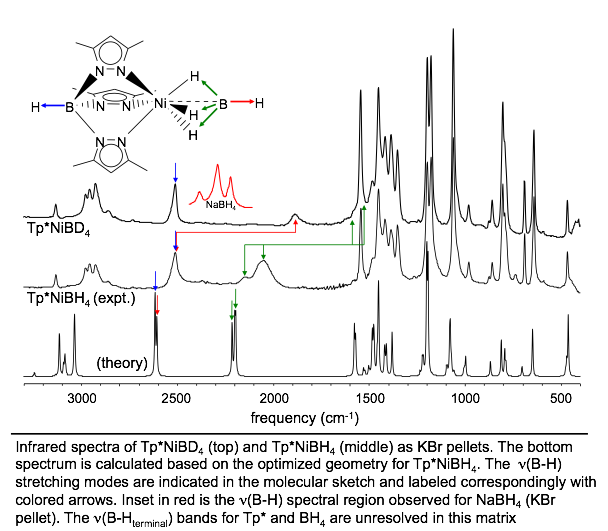
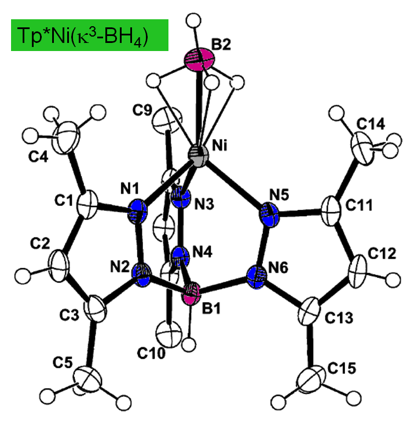
Stable nickel-borohydrides Inorganic Chemistry 2003,
42, 7945 - 7950 DOI: 10.1021/ic034687a A stable discrete nickel borohydride complex
(Tp*NiBH4 or Tp*NiBD4) was prepared using the nitrogen-donor
ligand hydrotris(3,5-dimethylpyrazolyl)borate (Tp*-). This complex represents one of the best
characterized nickel(II) borohydrides to date. Tp*NiBH4 and Tp*NiBD4 are stable toward air, boiling water, and high temperatures (mp
> 230 °C dec). X-ray
crystallographic measurements for Tp*NiBH4 showed a six-coordinate geometry for the complex, with the nickel(II) center facially
coordinated by three bridging hydrogen atoms from borohydride and a
tridentate Tp*- ligand. For Tp*NiBH4, the empirical formula is C15H26B2N6Ni, a
= 13.469(9)
Å, b = 7.740(1) Å, c = 18.851(2) Å, b = 107.605(9)°, the
space group is monoclinic P21/c,
and Z = 4. Infrared measurements confirmed the presence of bridging hydrogen atoms; both n(B-H)terminal and n(B-H)bridging are
assignable and shifted relative to n(B-D) of Tp*NiBD4 by amounts in agreement with theory. Despite their hydrolytic
stability, Tp*NiBH4 and Tp*NiBD4 readily reduce halocarbon substrates, leading to the complete series of Tp*NiX complexes (X =
Cl, Br, I). These reactions showed a pronounced
hydrogen/deuterium rate dependence (kH/kD ~ 3)
and sharp isosbestic points in progressive electronic spectra. Nickel K-edge X-ray
absorption spectroscopy (XAS) measurements of a hydride rich nickel center were obtained for Tp*NiBH4, Tp*NiBD4, and Tp*NiCl. X-ray absorption
near-edge spectroscopy results confirmed the similar six-coordinate geometries for Tp*NiBH4
and Tp*NiBD4. These contrasted with XAS results for the crystallographically characterized pseudotetrahedral
Tp*NiCl complex. The
stability of Tp*Ni-coordinated borohydride is significant given this ion’s
accelerated decomposition and hydrolysis in the presence of transition metals and simple metal salts. |
|
|
|
|
|
Reversible nickel-ammonia binding,
storage “Exchange equilibria of
variable nitrogen-donors at nickel(II)-scorpionates” Kristin
A. Thorvilson, Adeniyi Osinowo, Patrick J. Desrochers 239th American
Chemical Society National Meeting, San Francisco, CA, March 2010, INOR 230. Nickel(II) demonstrates a high affinity for
nitrogen-donor bases. Accordingly, Tp*Ni+ reversibly binds N-donors according to the
reaction: Tp*NiX + 3 N-donor à [Tp*Ni(N-donor)3]X,
where N-donor = imidazole, acetonitrile, or ammonia and X = Cl-,
Br-, I-, and BH4- and Tp* = the scorpionate
hydrotris(3,5-dimethylpyrazolyl)borate.
For all N-donors studied,
this reaction is exothermic, reflecting the exchange of stronger
nickel-N-donor for weaker nickel-X bonds. Variable temperature 11B
NMR of the Tp*NiX/acetonitrile
systems yielded thermodynamic parameters for these equilibria. We also describe reversible ammonia binding
at Tp*NiBH4, an interesting case because
the product, [Tp*Ni(NH3)3][BH4],
incorporates reactive hydridic B-H and protic N-H
groups in a single solid. This is reminiscent of magnesium
based hydrogen-storage materials incorporating a similar Mg-NH3—BH4
arrangement (Soloveichik,
et al. Inorg. Chem. 2008, p. 4290). These materials are expected to have applications
to solid-state ammonia storage and ammonia sensors. |
|